
As part of our rare disease round-up series, which provides up-to-date insights into the diagnosis and management of less commonly encountered dermatological conditions, we are delighted to feature a piece on the syndromic epidermal differentiation disorder (sEDD), SPINK5-sEDD (formerly known as Netherton syndrome).
 In this Q&A, Dr Pablo López Balboa, a paediatric dermatologist whose career has focused on advancing care for children with complex and rare skin diseases, shares his insights and expertise.
In this Q&A, Dr Pablo López Balboa, a paediatric dermatologist whose career has focused on advancing care for children with complex and rare skin diseases, shares his insights and expertise.
Originally trained in paediatrics in Spain, Dr López Balboa completed specialist training in paediatric dermatology at Niño Jesús University Children’s Hospital in Madrid in 2020. Since 2021, he has been part of the paediatric dermatology team at Great Ormond Street Hospital for Children, London, UK as a Senior Clinical Research Fellow, where his research focuses on stromal-cell therapy and drug repurposing in genodermatoses. His wider work includes research into biologic therapies, hair disorders and laser treatments.
Q. Could you give an overview of the clinical features and typical disease course of SPINK5-sEDD?
Netherton syndrome (NS), now referred to as SPINK5-sEDD under the new classification of syndromic epidermal differentiation disorders (sEDD), is a rare autosomal recessive skin disorder caused by loss-of-function mutations in the SPINK5 gene, which encodes a 15-domain serine protease inhibitor known as LEKTI (lymphoepithelial Kazal-type–related inhibitor).1,2
SPINK5-sEDD is characterized by a clinical triad comprising congenital ichthyosis, which may present as erythroderma (Netherton syndrome–congenital erythroderma, NS-CE) or ichthyosis linearis circumflexa (NS-ILC), trichorrhexis invaginata, and atopic manifestations with elevated serum IgE levels.3 Other features include multiple food allergies, linear desquamation along the edges of the palms, sparse hair and eyebrows, perineal confluent papillomatous lesions, eye disorders (such as conjunctivitis and ectropion), and various gastrointestinal complications, including gastro-oesophageal reflux, failure to thrive and diarrhoea.
The prevalence of SPINK5-sEDD is estimated at 1–9 per 1,000,000 individuals, with an incidence of approximately 1 in 200,000 births.4
Newborns with the condition typically present with neonatal scaling and exfoliative erythroderma, with annular scaling sometimes observed. Other common features include failure to thrive, a high risk of hypernatraemic dehydration, and alopecia, which occurs in around 30% of cases. Trichorrhexis invaginata may not be observed in the hair at birth but can often be detected in the newborn’s eyebrows. When approaching the differential diagnosis of neonatal erythroderma it is very important to follow the 6-step guideline outlined by Cuperus E. et al. in Figure 1.5
Figure 1: A 6-step approach of neonatal erythroderma5
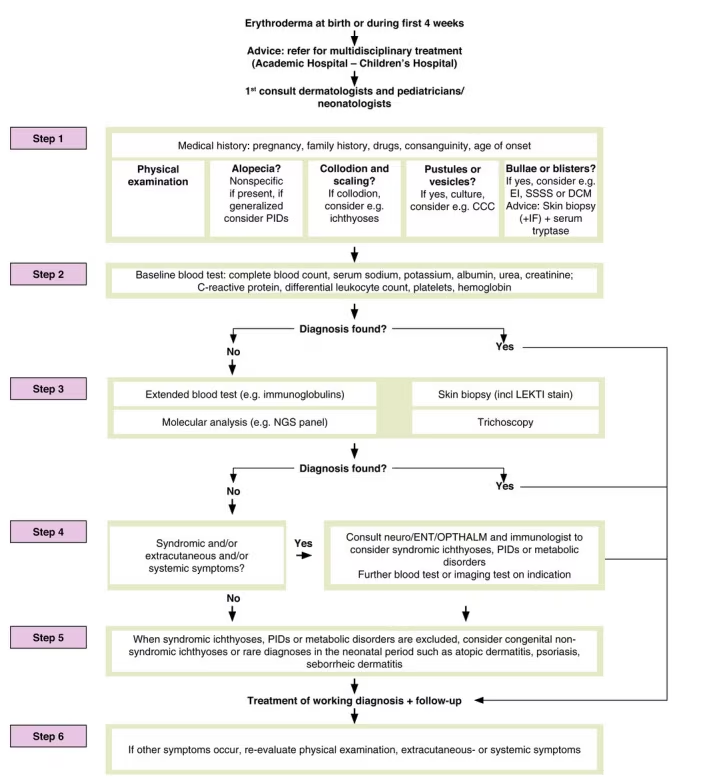
Reproduced with permission from Cuperus E et al. (2022) under a CC BY-NC-ND 4.0 attribution-noncommercial-noderivatives 4.0 international deed.5
A skin biopsy with LEKTI staining is the most reliable diagnostic method for patients with suspected SPINK5-sEDD. Histological findings may include compact parakeratosis with large nuclei, subcorneal or intracorneal splitting, absence of the stratum corneum, PAS-negative microabscesses, elongated rete ridges without suprapapillary thinning, dermal infiltrates rich in neutrophils and/or eosinophils and dilated blood vessels.6
Genetic testing can identify mutations in SPINK5; however, it is important to note that newer techniques such as next-generation sequencing (NGS) may miss mutations located outside the exons, such as intronic or other non-coding DNA variants. In such cases, whole-genome sequencing (WGS) is recommended.7,8
Q. What do we currently understand about the pathology of SPINK5-sEDD?
As discussed earlier, SPINK5-sEDD is driven by loss-of-function mutations in SPINK5, which encodes the serine protease inhibitor LEKTI. LEKTI normally restrains the activity of kallikrein-related peptidases (KLKs), a family of serine proteases that regulate epidermal desquamation. In the absence of functional LEKTI, KLKs become hyperactive in the upper granular and cornified layers of the epidermis, initiating a cascade of pathological events.9,10
Protease cascade
Matriptase and KLK5 act as central initiators of a proteolytic cascade, activating additional proteases such as KLK7, KLK14, and neutrophil elastase-2 (ELA2). This unregulated protease activity contributes to skin pathology through several mechanisms:
-
Skin barrier dysfunction: Excessive cleavage of key structural and enzymatic proteins undermines epidermal integrity. These include corneodesmosomal cadherins (desmoglein-1 [DSG1] and desmocollin-1 [DSC1]), corneodesmosin (CDSN), and premature degradation of filaggrin (FLG) and its precursor pro-FLG. Lipid-processing enzymes such as β-glucocerebrosidase (β-GlcCer’ase) and acidic sphingomyelinase (aSMase) are also targeted, further weakening the stratum corneum.
-
Hair shaft abnormalities: Aberrant degradation of desmoglein-3 (DSG3) and desmoglein-4 (DSG4) compromises hair anchoring and structure, accounting for the characteristic trichorrhexis invaginata.
-
Inflammation: KLK-driven proteolytic activation of the proteinase-activated receptor-2 (PAR-2) on keratinocytes induces the synthesis of pro-inflammatory cytokines. Additional pro-inflammatory stimuli include KLK-mediated activation of pro-interleukin-1β (pro-IL-1β) and altered processing of antimicrobial peptides such as LL-37.
-
Pruritus: The kallikrein-related peptidases, KLK5, KLK7, and KLK14, activate PAR-2 receptors on sensory nerves, which directly contributes to chronic itch.
Amplification loops and innate sensing
Skin barrier breakdown exposes the epidermis to allergens and microbes, such as Staphylococcus aureus. Recognition of pathogen-associated molecular patterns (PAMPs) by Toll-like receptors (TLRs) and NOD-like receptors (NLRs) in keratinocytes amplifies the inflammatory response. In addition, alarmins such as IL-33, S100A8, and S100A9 are released in response to cellular stress or tissue damage, further driving cytokine production.
Immune crosstalk and chronic inflammation
Pro-inflammatory mediators secreted by keratinocytes, including IL-1β, IL-17C, IL-36, IL-6, IL-8, TNF-α, CCL20, CXCL5, and thymic stromal lymphopoietin (TSLP), drive recruitment and activation of immune cells. This results in infiltration of Th2 cells, Th17 cells, neutrophils, eosinophils and mast cells into the dermis.
-
Th2 cells release IL-4, IL-13, and IL-31, promoting IgE class switching and pruritus.
-
Th17 cells produce IL-17A, IL-22, and IL-36, which exacerbate epidermal hyperplasia and inflammation.
-
Mast cells and eosinophils contribute through the release of histamine, tryptase, chemokines, and proteases that sustain inflammation and itch.
-
Neutrophils secrete proteases and defensins that aggravate tissue injury.
Together, this creates a self-perpetuating vicious cycle in which barrier dysfunction facilitates antigen entry and immune activation, while inflammation further compromises epidermal structure and function (Figure 2).9
Figure 2: Signalling pathways underlying the pathophysiology of Netherton syndrome.9
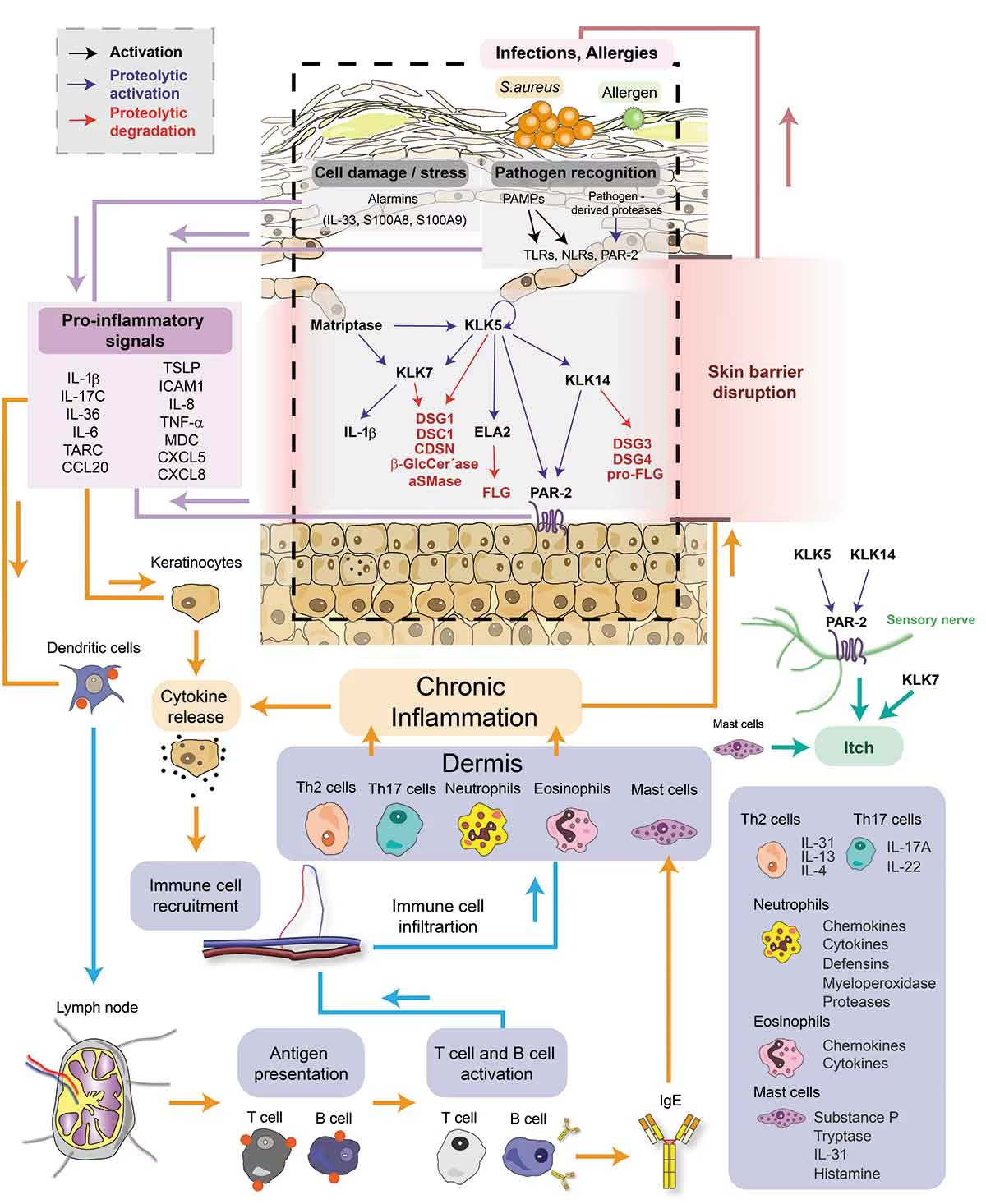
Reproduced with permission from Petrova E & Hovnanian A (2020) under a CC BY-NC-ND 4.0 attribution-noncommercial-noderivatives 4.0 international deed.9
Emerging insights: IL-17/IL-36 pathways
Barbieux C et al. previously demonstrated that the IL-17/IL-36 signature constitutes a shared feature of congenital erythroderma (CE) and ichthyosis linearis circumflexa (ILC).11 The study further distinguished these conditions based on their immune profiles, with a Th2/complement-driven axis in ILC and a type I IFN/Th9-driven axis in CE. Recent data from Petrova E et al. highlights the KLK/IL-36 pathway as an additional driver of pathology in SPINK5-sEDD. In this pathway, IL-36 cytokines, activated by KLKs, promote keratinocyte inflammation and Th17 polarization, positioning this axis as a promising therapeutic target.12
Q. What are the key complications that dermatologists should be aware of when treating patients with SPINK5-sEDD?
The main systemic and cutaneous complications that influence both morbidity and mortality in patients with SPINK5-sEDD are:3,13
-
Severe skin barrier dysfunction: Severe skin barrier dysfunction can lead to dehydration and hypernatremia in the neonatal period. More than 50% of affected newborns may experience hypernatremic dehydration. Chronic features include xerosis, ichthyosiform erythroderma, scaling, fissuring. It is important to highlight the need for caution with any topical treatments with active ingredients, like topical corticosteroids, calcineurin inhibitors or keratolytics (e.g. urea, salicylic acid or lactic acid).
-
Skin infections: There is a severe risk of septicaemia in newborns with SPINK5-sEDD, which is one of the main causes of mortality. Due to skin barrier dysfunction, affected individuals have a high frequency of bacterial, viral and fungal skin infections. Papillomatous lesions have also been described, typically non-malignant and most often affecting the perineal area, which can, in severe cases, resemble a Buschke–Lowenstein tumour caused by HPV.14
-
Allergies: Because of the severely impaired skin barrier, patients with SPINK5-sEDD are at high risk of developing food allergies through cutaneous sensitization. Early skin contact with food proteins, prior to being ingested, should be avoided, as this may predispose to severe food allergies. Given the risk of systemic reactions, including anaphylaxis, affected patients may require prescription and regular carriage of an epinephrine autoinjector.15
-
Failure to thrive and growth retardation: As a result of increased transepidermal water and protein loss, chronic inflammation, and recurrent infections, individuals with SPINK5-sEDD may fail to thrive and grow as expected.
-
Hair abnormalities: Individuals with SPINK5-sEDD may present with hair abnormalities, including trichorrhexis invaginata (“bamboo hair”), pili torti, and trichorrhexis nodosa. These features can aid in diagnosis but may also contribute to cosmetic and psychosocial concerns.
-
Atopic diathesis: Many affected individuals have a predisposition to allergic conditions, characterised by a severe atopic dermatitis–like phenotype, allergic rhinitis, asthma, and food allergies, with an associated risk of anaphylaxis.
-
Severe pruritus: Severe itching from the condition can significantly contribute to reduced quality of life, can become debilitating and may even interfere with sleep.
-
Increased risk of cutaneous malignancy: Some patients with longstanding disease may develop squamous cell carcinoma, potentially as a consequence of chronic inflammation and persistent barrier dysfunction.
-
Psychosocial burden: SPINK5-sEDD is a chronic, visible skin condition characterised by hair abnormalities, scaly skin, erythema, and pruritus, which frequently leads to reduced self-esteem, anxiety and depression.
Q. What advice would you give dermatologists to help distinguish SPINK5-sEDD from more common skin conditions?
When evaluating a newborn presenting with erythroderma and scaly skin, it is important to follow the 6-step guidelines mentioned previously (Figure 1). Skin biopsy and hair microscopy or trichoscopy of the scalp or eyebrows can be very useful in identifying the characteristic triad of SPINK5-sEDD: scaly erythroderma or ichthyosis linearis circumflexa, trichorrhexis invaginata, and atopic manifestations.5
In addition, atopic manifestations (such as eczema, elevated IgE levels, and food allergies) may be present and are often disproportionate to those seen in typical atopic dermatitis. However when diagnostic uncertainty remains, a skin biopsy with LEKTI staining is essential to confirm or exclude the diagnosis, and/or genetic testing should be performed to identify SPINK5 mutations.
Q. What are the current approaches to managing SPINK5-sEDD?
 The management of SPINK5-sEDD requires a multidisciplinary approach involving a paediatrician, dermatologist, allergist, dietitian, ENT specialist and ophthalmologist, as well as occupational therapists, social workers and psychologists. Below, I outline key considerations for managing patients with this condition; however, two key publications by Mazereeuw-Hautier et al. and Barbati et al. also provide excellent guidance on its management.16,17
The management of SPINK5-sEDD requires a multidisciplinary approach involving a paediatrician, dermatologist, allergist, dietitian, ENT specialist and ophthalmologist, as well as occupational therapists, social workers and psychologists. Below, I outline key considerations for managing patients with this condition; however, two key publications by Mazereeuw-Hautier et al. and Barbati et al. also provide excellent guidance on its management.16,17
Nutrition: Nutrition plays a key role in the management of patients living with SPINK5-sEDD. Growth difficulties are common due to their often hypercatabolic state, potential malabsorption, and the need for vitamin D supplementation. During the first months of life, patients experience transepidermal water loss (TEWL) of up to 45.1 g/m²/h, meaning fluid requirements may reach up to 250 mL/kg/day during the first 6 months. SPINK5-sEDD can also be associated with eosinophilic oesophagitis.15
Allergies: In a study of 21 patients with SPINK5-sEDD, 84.2% presented with IgE-mediated food allergies, ranging from 1 to 12 food allergies per patient.15 Due to markedly elevated IgE levels, specific IgE allergy testing may yield false-positive results; therefore, a low threshold for hospital-based oral food challenges is recommended. With an appropriate dietetic plan and close follow-up, it is advisable to initiate weaning early to reduce the risk of future food allergies. Food-based moisturisers or emollients should be avoided on the skin until the corresponding food has been safely introduced orally.
Dermatology: Regular use of topical antiseptics (2–3 times per week) is recommended to cleanse the skin and improve the microbiome, reducing colonisation by fungi and bacteria such as Staphylococcus aureus. Liberal use of emollients and keratolytics is also encouraged according to patient need. Topical corticosteroids and calcineurin inhibitors can be used, but due to increased absorption and risk of systemic side effects, their use should be restricted to short-term flare management on small areas. Oral retinoids are not recommended, as they may increase skin fragility. Narrowband UVB (NB-UVB) phototherapy is not routinely recommended, although a few selected case reports reported minimal benefit. Long-term phototherapy should be avoided due to an increased risk of skin cancer.18
Biologic treatments: As described previously, multiple interleukins play a role in SPINK5-sEDD, with particular interest in the Th17 pathway. A large recent retrospective, observational, international, multicentre study of 98 patients with congenital ichthyosis (CI), including 29 with SPINK5-sEDD, showed that approximately 50% of CI patients respond to biologic treatments. SPINK5-sEDD appeared to respond particularly well, with no major safety concerns reported. The biologics most frequently used in SPINK5-sEDD are dupilumab (an anti-IL-4Rα, blocking IL-4/IL-13) and ixekizumab/secukinumab (an anti-IL-17A). Multiple case reports have demonstrated clinical improvement, with response times of up to 6 months. No serious adverse events have been reported, and younger patients tend to respond better (the youngest reported treated with anti-IL17A was 2 years and 11 months old child). Clinical response appears to be dose dependent. Dupilumab seems to improve itch while anti-IL17A improves erythroderma. New studies are starting to combine both treatments showing increased response to treatment without any severe adverse events.19–22
Data on long-term follow-up remain limited. Evidence for JAK inhibitors in SPINK5-sEDD is scarce; available case reports show only transient benefit, so there is currently insufficient evidence to recommend their use.
Q. What emerging treatments or strategies show the most promise for improving patient outcomes?
Promising new treatments: Ongoing research is investigating kallikrein inhibitors targeting KLK5, KLK7, and KLK14, which have shown encouraging results in preclinical studies, with clinical data in patients still awaited.22
Additionally, zinc has been shown to reversibly inhibit KLK5, KLK7, KLK8 and KLK14, whereas an acidic pH activates these enzymes. A single case report described clinical improvement in a newborn treated with an ointment containing 40% zinc oxide and sodium carbonate.23
Gene therapy: No gene therapy is currently available. A phase 1 trial using gene-modified autologous epidermal sheets demonstrated transient LEKTI expression, suggesting that targeting keratinocyte stem cells may be required to achieve sustained LEKTI production.24
Cite: López Balboa P. SPINK5-sEDD: Pathophysiology, clinical features, diagnosis and management. touchDERMATOLOGY. 26 November, 2025.



